Download updated gene description (copied from “2018-10-01-…Rmd”)
Most updated annotation can be downloaded from TAIR website (for subscribers). I used “TAIR Data 20180630”.
library(tidyverse);library(readxl);library(readr)## ── Attaching packages ─────────────────────────────────────── tidyverse 1.3.0 ──## ✓ ggplot2 3.3.3 ✓ purrr 0.3.4
## ✓ tibble 3.0.4 ✓ dplyr 1.0.2
## ✓ tidyr 1.1.2 ✓ stringr 1.4.0
## ✓ readr 1.4.0 ✓ forcats 0.5.0## ── Conflicts ────────────────────────────────────────── tidyverse_conflicts() ──
## x dplyr::filter() masks stats::filter()
## x dplyr::lag() masks stats::lag()# turn on TAIR subsciption before this procedure
At.gene.name <-read_tsv("https://www.arabidopsis.org/download_files/Subscriber_Data_Releases/TAIR_Data_20180630/gene_aliases_20180702.txt.gz") # Does work from home when I use Pulse Secure.
# How to concatenate multiple symbols in the same AGI
At.gene.name <- At.gene.name %>% group_by(name) %>% summarise(symbol2=paste(symbol,collapse=";"),full_name=paste(full_name,collapse=";"))
At.gene.name %>% dplyr::slice(100:110) # OK
# download cDNA info directly from TAIR
At_cdna<-Biostrings::readDNAStringSet("https://www.arabidopsis.org/download_files/Sequences/TAIR10_blastsets/TAIR10_cdna_20110103_representative_gene_model_updated")
At_cdna # 33602 genesusing downloaded file
system("gunzip -c ../../../resources/2018-09-26-over-representation-analysis-1-goseq-with-arabidopsis-go-term_files/TAIR10_cdna_20110103_representative_gene_model_updated.gz > ../../../resources/2018-09-26-over-representation-analysis-1-goseq-with-arabidopsis-go-term_files/TAIR10_cdna_20110103_representative_gene_model_updated")
At_cdna <- Biostrings::readDNAStringSet(file.path("..","..","..","resources","2018-09-26-over-representation-analysis-1-goseq-with-arabidopsis-go-term_files","TAIR10_cdna_20110103_representative_gene_model_updated"))
At_cdna## A DNAStringSet instance of length 33602
## width seq names
## [1] 2394 AGAAAACAGTCGACCGTCATT...TATTTGGTAATTTTTTGAGTC AT1G50920.1 | Sym...
## [2] 546 ATGACTCGTTTGTTGCCTTAC...CAAGTTGATTCTGGTACATAG AT1G36960.1 | Sym...
## [3] 2314 ATGGATTCAGAGTCAGAGTCA...CCTGGTGCATTGTGTTTCTCC AT1G44020.1 | Sym...
## [4] 1658 TCGTTTCGTCGTCGATCAGAA...ATCGATTACATGCTACATTTT AT1G15970.1 | Sym...
## [5] 1453 ATTGAAAAGAAAACACATCCC...GACCACCAAAATCTTCTCATA AT1G73440.1 | Sym...
## ... ... ...
## [33598] 87 GGATGGATGTCTGAGCGGTTG...GTTCGAATCCCTCTCCATCCG ATMG00420.1 | Sym...
## [33599] 384 ATGCTCCCCGCCGGTTGTTGG...CTACGATACTTAACTATATAA ATMG01330.1 | Sym...
## [33600] 573 ATGGATAACCAATTCATTTTC...GAACAGCGTAGCGACGGATAA ATMG00070.1 | Sym...
## [33601] 366 ATGGCATCAAAAATCCGCAAA...GATCCTTCTGCATACGCATAA ATMG00130.1 | Sym...
## [33602] 74 GCGCTCTTAGTTCAGTTCGGT...GTTCAAATCCTACAGAGCGTG ATMG00930.1 | Sym...system("rm ../../../resources/2018-09-26-over-representation-analysis-1-goseq-with-arabidopsis-go-term_files/TAIR10_cdna_20110103_representative_gene_model_updated")Updated hormone responsive gene list1
hormone.responsiveness6.DF.s.list2.DF2<-readxl::read_xlsx(file.path("..","..","..","resources","2018-10-10-over-representation-analysis-2-goseq-using-custom-categories_files","Supplemental_Dataset3_source_of_custom_categories.xlsx"),skip=15,sheet=1) # Does not work. why?
# because uglyURLs and canonifyURLs = true on 100818. I commented out them.
# Ask Kazu for this xlsx file.
hormone.responsiveness6.DF.s.list2.DF2## # A tibble: 519 x 20
## ABAdown ACCup ACCdown BLup BLdown IAAup IAAdown MJup MJdown CKup CKdown
## <chr> <chr> <chr> <chr> <chr> <chr> <chr> <chr> <chr> <chr> <chr>
## 1 AT1G21… AT1G… AT1G70… AT1G… AT1G2… AT1G… AT1G74… AT1G… AT1G7… AT1G… AT1G4…
## 2 AT1G21… AT1G… AT1G29… AT1G… AT1G1… AT1G… AT1G31… AT1G… AT1G3… AT1G… AT1G7…
## 3 AT1G49… AT1G… AT1G28… AT1G… AT1G6… AT1G… AT1G17… AT1G… AT1G6… AT1G… AT1G7…
## 4 AT1G73… AT1G… AT1G64… AT2G… AT1G0… AT1G… AT1G13… AT1G… AT1G7… AT1G… AT2G4…
## 5 AT1G70… AT1G… AT1G72… AT2G… AT1G7… AT1G… AT1G18… AT1G… AT1G4… AT1G… AT2G3…
## 6 AT1G31… AT2G… AT2G26… AT2G… AT1G1… AT1G… AT1G31… AT1G… AT1G7… AT1G… AT2G3…
## 7 AT1G77… AT2G… AT2G24… AT2G… AT1G7… AT1G… AT1G77… AT1G… AT1G1… AT1G… AT2G2…
## 8 AT1G16… AT2G… AT2G03… AT2G… AT2G2… AT1G… AT1G13… AT1G… AT1G6… AT1G… AT4G1…
## 9 AT1G70… AT2G… AT3G13… AT2G… AT2G3… AT1G… AT1G72… AT1G… AT1G1… AT1G… AT4G3…
## 10 AT1G77… AT2G… AT3G56… AT3G… AT2G1… AT1G… AT1G72… AT1G… AT1G6… AT1G… AT4G3…
## # … with 509 more rows, and 9 more variables: GAup <chr>, GAdown <chr>,
## # PIFtarget <chr>, MYC234up <chr>, MYC234down <chr>, SAup <chr>,
## # Sadown <chr>, WRKY33up <chr>, WRKY33down <chr># conver to list obejct compatible with goseq() belowformat hormone.responsiveness6.DF.s.list2.DF2 into goseq() compatible list (run once)
# extract AGI from At_cdna
At.gene.name<-tibble(name=names(At_cdna)) %>% separate(name,into=c("name2","Symbol","description"),sep=" \\|",extra="drop",fill="left") %>% mutate(AGI=str_remove(name2,pattern="\\.[[:digit:]]+"))
# make presence/absense hormone responsive gene table (dataframe)
for(i in 1:20) {
genes<-hormone.responsiveness6.DF.s.list2.DF2[!is.na(hormone.responsiveness6.DF.s.list2.DF2[,i]),i] %>% as_vector()
temp<-data.frame(AGI=genes,category=names(hormone.responsiveness6.DF.s.list2.DF2)[i])
#At.gene.name %>% left_join(temp,by=c("name"="AGI")) -> At.gene.name
At.gene.name %>% left_join(temp,by="AGI") -> At.gene.name
}
names(At.gene.name)[5:24] <- names(hormone.responsiveness6.DF.s.list2.DF2)
At.gene.name %>% filter(str_detect(AGI,pattern="AT1G|AT2G|AT3G|AT4G|AT5G|ATC|ATM")) %>% unite(category,5:24,sep=",")->test2
test2[1:10,] %>% mutate(category=gsub("NA","",category)) %>% mutate(category=gsub(",","",category))
# only select genes with AGI name and concatenate categories
At.gene.name %>% filter(str_detect(AGI,pattern="AT1G|AT2G|AT3G|AT4G|AT5G|ATC|ATM")) %>% unite(category,5:24,sep=",") %>% mutate(category=gsub("NA,","",category)) %>% mutate(category=gsub(",NA","",category)) %>% mutate(category=gsub("NA","",category)) ->test3
# convert into list object
temp.list<-list()
for(i in 1:dim(test3)[1]) {
temp.list[[i]]<-test3 %>% slice(i) %>% pull(category)
}
names(temp.list)<-test3 %>% pull(AGI)
# split concatanated categories in each gene
At.hormone.responsive.list<-lapply(temp.list,function(x) unlist(strsplit(x, split=",")))
# save
save(At.hormone.responsive.list,file=file.path("..","..","..","resources","2018-10-10-over-representation-analysis-2-goseq-using-custom-categories_files","At.hormone.responsive.list.Rdata"))GOseq ORA function with Arabidopss thaliana hormone responsive genes (under construction)
load(file.path("..","..","..","resources","2018-10-10-over-representation-analysis-2-goseq-using-custom-categories_files","At.hormone.responsive.list.Rdata"))
head(At.hormone.responsive.list)## $AT1G50920
## character(0)
##
## $AT1G36960
## character(0)
##
## $AT1G44020
## character(0)
##
## $AT1G15970
## character(0)
##
## $AT1G73440
## character(0)
##
## $AT1G75120
## character(0)library(ShortRead);library(goseq);library(GO.db);library("annotate")## Warning: package 'S4Vectors' was built under R version 3.6.3## Warning: package 'GenomeInfoDb' was built under R version 3.6.3## Warning: package 'DelayedArray' was built under R version 3.6.3# bias.data vector must have the same length as DEgenes vector!
## if you want to test expressed genes as background, add background in this function
GOseq.At.customcat.ORA<-function(genelist,padjust=0.05,custom.category.list=At.hormone.responsive.list,cdna=At_cdna,background="") { # return GO enrichment table, padjus, padjust=0.05. New BLAST2GO results with only BP Brgo.v2.5.BP.list is used (042518). Brgo.v2.5.BP.list is based on Hajar's Blast2go, which should be replaced with Brgo.Atgoslim.BP.list and giving a new name to this function (080818). cdna is DNAstring object. background is a vector of genes.
#bias<-nchar(cdna)
bias<-Biostrings::width(cdna)
# add name to "bias". cdna
#names(bias)<-names(cdna)
names(bias) <- tibble(name=names(cdna)) %>% separate(name,into=c("name2","Symbol","description"),sep=" \\|",extra="drop",fill="left") %>% mutate(AGI=str_remove(name2,pattern="\\.[[:digit:]]+")) %>% pull(AGI)
if(background=="") {print("Use all genes in genome as background")} else {
# select only expressed geens
bias <- bias[background]
print("Expessed genes are used as background.")
print(paste("length of bias is ",length(bias)))
}
TF<-(names(bias) %in% genelist)*1
names(TF)<-names(bias)
#print(TF)
pwf<-nullp(TF,bias.data=bias)
#print(pwf$DEgenes)
GO.pval <- goseq(pwf,gene2cat=custom.category.list,use_genes_without_cat=TRUE) # format became different in new goseq version (021111). Does not work (042716)
# calculate p ajust by BH
GO.pval$over_represented_padjust<-p.adjust(GO.pval$over_represented_pvalue,method="BH")
if(GO.pval$over_represented_padjust[1]>padjust) return("no enriched GO")
else {
enriched.GO<-GO.pval[GO.pval$over_represented_padjust<padjust,]
print("enriched.GO is")
print(enriched.GO)
return(enriched.GO)
}
}test the function
load(file.path("..","..","..","resources","2018-10-10-over-representation-analysis-2-goseq-using-custom-categories_files","dge.nolow.Rdata"))
expressed.genes<-gsub("(.)(\\.[[:digit:]]+)","\\1",as_vector(dge.nolow$genes))## Loading required package: edgeR## Loading required package: limma##
## Attaching package: 'limma'## The following object is masked from 'package:BiocGenerics':
##
## plotMAGOseq.At.customcat.ORA(genelist=hormone.responsiveness6.DF.s.list2.DF2$IAAup, background="") # Total genes in genome are background## [1] "Use all genes in genome as background"## Warning in pcls(G): initial point very close to some inequality constraints## Using manually entered categories.## Calculating the p-values...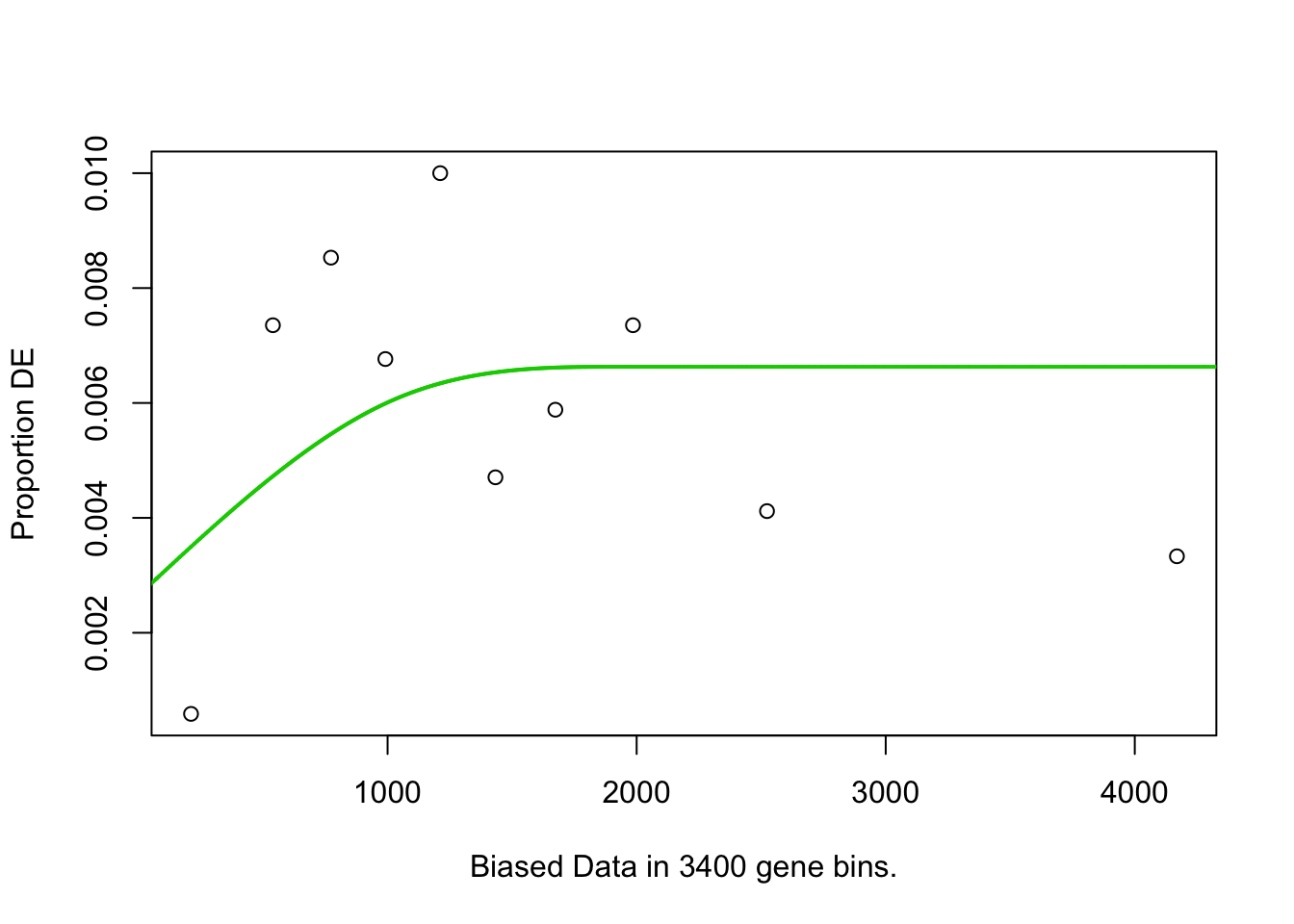
## [1] "enriched.GO is"
## category over_represented_pvalue under_represented_pvalue numDEInCat
## 11 IAAup 0.000000e+00 1.0000000 198
## 12 MJdown 4.395205e-21 1.0000000 23
## 5 BLup 7.235275e-18 1.0000000 11
## 19 WRKY33down 1.763824e-16 1.0000000 18
## 16 PIFtarget 2.001183e-14 1.0000000 11
## 3 ACCup 2.464518e-11 1.0000000 8
## 13 MJup 6.723578e-09 1.0000000 18
## 1 ABAdown 1.782286e-07 1.0000000 12
## 4 BLdown 2.043251e-06 0.9999999 5
## 18 SAup 8.089212e-06 0.9999992 8
## 8 GAdown 1.526185e-05 0.9999993 5
## 7 CKup 5.622343e-04 0.9999614 4
## 10 IAAdown 6.516900e-03 0.9994291 3
## numInCat over_represented_padjust
## 11 198 0.000000e+00
## 12 230 4.395205e-20
## 5 28 4.823517e-17
## 19 195 8.819122e-16
## 16 52 8.004731e-14
## 3 34 8.215059e-11
## 13 519 1.921022e-08
## 1 269 4.455716e-07
## 4 33 4.540558e-06
## 18 168 1.617842e-05
## 8 50 2.774883e-05
## 7 60 9.370572e-04
## 10 61 1.002600e-02## category over_represented_pvalue under_represented_pvalue numDEInCat
## 11 IAAup 0.000000e+00 1.0000000 198
## 12 MJdown 4.395205e-21 1.0000000 23
## 5 BLup 7.235275e-18 1.0000000 11
## 19 WRKY33down 1.763824e-16 1.0000000 18
## 16 PIFtarget 2.001183e-14 1.0000000 11
## 3 ACCup 2.464518e-11 1.0000000 8
## 13 MJup 6.723578e-09 1.0000000 18
## 1 ABAdown 1.782286e-07 1.0000000 12
## 4 BLdown 2.043251e-06 0.9999999 5
## 18 SAup 8.089212e-06 0.9999992 8
## 8 GAdown 1.526185e-05 0.9999993 5
## 7 CKup 5.622343e-04 0.9999614 4
## 10 IAAdown 6.516900e-03 0.9994291 3
## numInCat over_represented_padjust
## 11 198 0.000000e+00
## 12 230 4.395205e-20
## 5 28 4.823517e-17
## 19 195 8.819122e-16
## 16 52 8.004731e-14
## 3 34 8.215059e-11
## 13 519 1.921022e-08
## 1 269 4.455716e-07
## 4 33 4.540558e-06
## 18 168 1.617842e-05
## 8 50 2.774883e-05
## 7 60 9.370572e-04
## 10 61 1.002600e-02GOseq.At.customcat.ORA(genelist=hormone.responsiveness6.DF.s.list2.DF2$IAAup, background=expressed.genes) # Expressed genes in samples used for DEGs are background.## Warning in if (background == "") {: the condition has length > 1 and only the
## first element will be used## [1] "Expessed genes are used as background."
## [1] "length of bias is 16799"## Using manually entered categories.
## Calculating the p-values...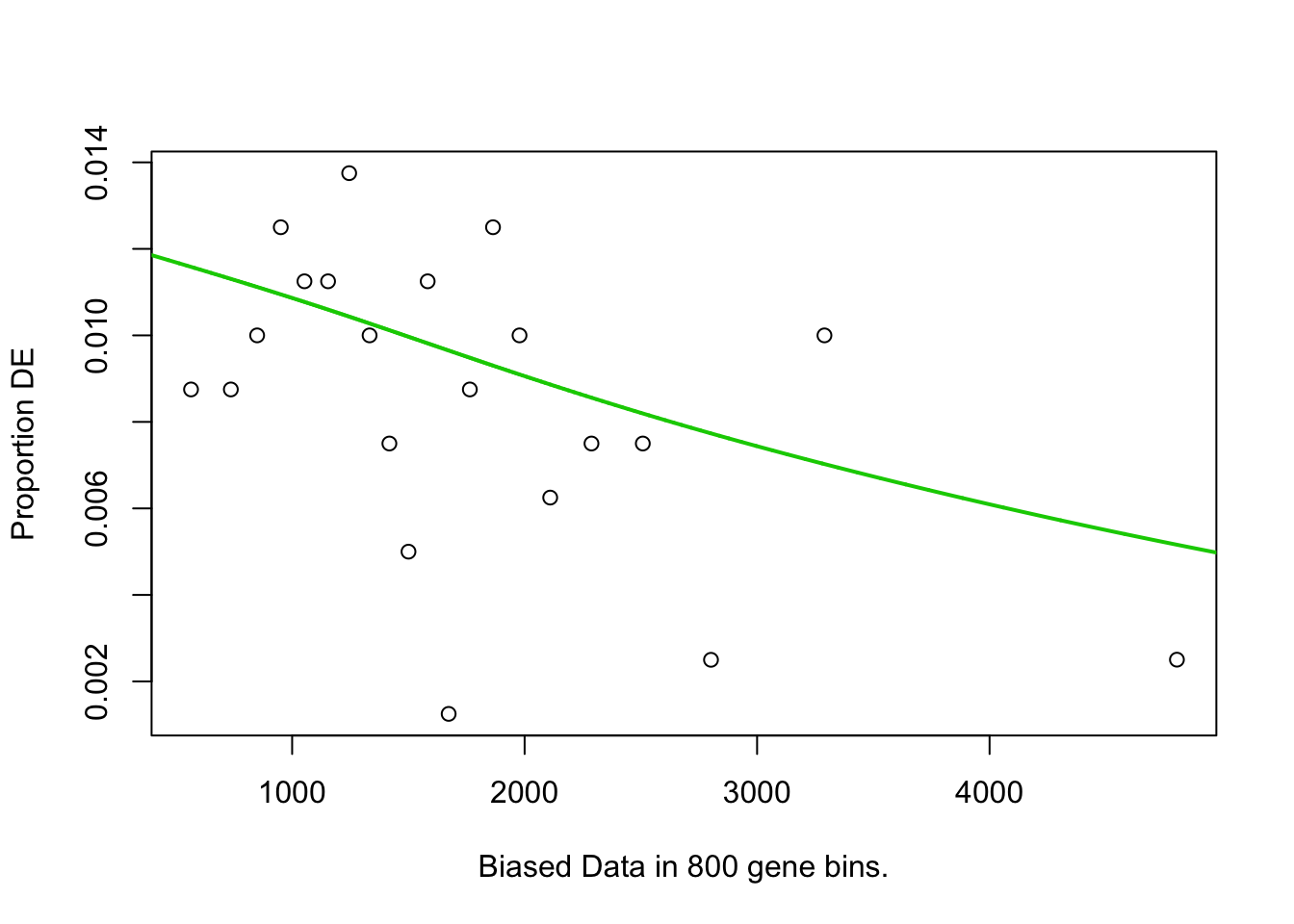
## [1] "enriched.GO is"
## category over_represented_pvalue under_represented_pvalue numDEInCat
## 11 IAAup 0.000000e+00 1.0000000 143
## 12 MJdown 5.092312e-16 1.0000000 21
## 5 BLup 6.670465e-13 1.0000000 9
## 19 WRKY33down 1.372204e-12 1.0000000 16
## 16 PIFtarget 4.126366e-10 1.0000000 8
## 3 ACCup 3.002866e-06 0.9999999 5
## 4 BLdown 8.892919e-06 0.9999997 5
## 13 MJup 4.273610e-05 0.9999900 14
## 1 ABAdown 7.261961e-05 0.9999870 10
## 18 SAup 9.374241e-05 0.9999867 8
## 8 GAdown 5.425846e-04 0.9999639 4
## 7 CKup 7.477068e-03 0.9993214 3
## numInCat over_represented_padjust
## 11 143 0.000000e+00
## 12 221 5.092312e-15
## 5 25 4.446976e-12
## 19 159 6.861019e-12
## 16 34 1.650546e-09
## 3 26 1.000955e-05
## 4 33 2.540834e-05
## 13 454 1.068403e-04
## 1 257 1.613769e-04
## 18 154 1.874848e-04
## 8 43 9.865175e-04
## 7 46 1.246178e-02## category over_represented_pvalue under_represented_pvalue numDEInCat
## 11 IAAup 0.000000e+00 1.0000000 143
## 12 MJdown 5.092312e-16 1.0000000 21
## 5 BLup 6.670465e-13 1.0000000 9
## 19 WRKY33down 1.372204e-12 1.0000000 16
## 16 PIFtarget 4.126366e-10 1.0000000 8
## 3 ACCup 3.002866e-06 0.9999999 5
## 4 BLdown 8.892919e-06 0.9999997 5
## 13 MJup 4.273610e-05 0.9999900 14
## 1 ABAdown 7.261961e-05 0.9999870 10
## 18 SAup 9.374241e-05 0.9999867 8
## 8 GAdown 5.425846e-04 0.9999639 4
## 7 CKup 7.477068e-03 0.9993214 3
## numInCat over_represented_padjust
## 11 143 0.000000e+00
## 12 221 5.092312e-15
## 5 25 4.446976e-12
## 19 159 6.861019e-12
## 16 34 1.650546e-09
## 3 26 1.000955e-05
## 4 33 2.540834e-05
## 13 454 1.068403e-04
## 1 257 1.613769e-04
## 18 154 1.874848e-04
## 8 43 9.865175e-04
## 7 46 1.246178e-02Session info
sessionInfo()## R version 3.6.2 (2019-12-12)
## Platform: x86_64-apple-darwin15.6.0 (64-bit)
## Running under: macOS Mojave 10.14.6
##
## Matrix products: default
## BLAS: /Library/Frameworks/R.framework/Versions/3.6/Resources/lib/libRblas.0.dylib
## LAPACK: /Library/Frameworks/R.framework/Versions/3.6/Resources/lib/libRlapack.dylib
##
## locale:
## [1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
##
## attached base packages:
## [1] stats4 parallel stats graphics grDevices utils datasets
## [8] methods base
##
## other attached packages:
## [1] edgeR_3.28.1 limma_3.42.2
## [3] annotate_1.64.0 XML_3.99-0.3
## [5] GO.db_3.10.0 AnnotationDbi_1.48.0
## [7] goseq_1.38.0 geneLenDataBase_1.22.0
## [9] BiasedUrn_1.07 ShortRead_1.44.3
## [11] GenomicAlignments_1.22.1 SummarizedExperiment_1.16.1
## [13] DelayedArray_0.12.3 matrixStats_0.57.0
## [15] Biobase_2.46.0 Rsamtools_2.2.3
## [17] GenomicRanges_1.38.0 GenomeInfoDb_1.22.1
## [19] Biostrings_2.54.0 XVector_0.26.0
## [21] IRanges_2.20.2 S4Vectors_0.24.4
## [23] BiocParallel_1.20.1 BiocGenerics_0.32.0
## [25] readxl_1.3.1 forcats_0.5.0
## [27] stringr_1.4.0 dplyr_1.0.2
## [29] purrr_0.3.4 readr_1.4.0
## [31] tidyr_1.1.2 tibble_3.0.4
## [33] ggplot2_3.3.3 tidyverse_1.3.0
##
## loaded via a namespace (and not attached):
## [1] colorspace_2.0-0 hwriter_1.3.2 ellipsis_0.3.1
## [4] fs_1.5.0 rstudioapi_0.13 bit64_4.0.5
## [7] fansi_0.4.1 lubridate_1.7.9.2 xml2_1.3.2
## [10] splines_3.6.2 knitr_1.31 jsonlite_1.7.2
## [13] broom_0.7.3 dbplyr_2.0.0 png_0.1-7
## [16] compiler_3.6.2 httr_1.4.2 backports_1.2.1
## [19] assertthat_0.2.1 Matrix_1.3-2 cli_2.2.0
## [22] htmltools_0.5.1.1 prettyunits_1.1.1 tools_3.6.2
## [25] gtable_0.3.0 glue_1.4.2 GenomeInfoDbData_1.2.2
## [28] rappdirs_0.3.1 Rcpp_1.0.6 cellranger_1.1.0
## [31] vctrs_0.3.6 nlme_3.1-151 blogdown_1.2.2
## [34] rtracklayer_1.46.0 xfun_0.22 rvest_0.3.6
## [37] lifecycle_0.2.0 zlibbioc_1.32.0 scales_1.1.1
## [40] hms_0.5.3 RColorBrewer_1.1-2 yaml_2.2.1
## [43] curl_4.3 memoise_1.1.0 biomaRt_2.42.1
## [46] latticeExtra_0.6-29 stringi_1.5.3 RSQLite_2.2.1
## [49] highr_0.8 GenomicFeatures_1.38.2 rlang_0.4.10
## [52] pkgconfig_2.0.3 bitops_1.0-6 evaluate_0.14
## [55] lattice_0.20-41 bit_4.0.4 tidyselect_1.1.0
## [58] magrittr_2.0.1 bookdown_0.21 R6_2.5.0
## [61] generics_0.1.0 DBI_1.1.0 pillar_1.4.7
## [64] haven_2.3.1 withr_2.3.0 mgcv_1.8-33
## [67] RCurl_1.98-1.2 modelr_0.1.8 crayon_1.3.4
## [70] utf8_1.1.4 BiocFileCache_1.10.2 rmarkdown_2.7
## [73] jpeg_0.1-8.1 progress_1.2.2 locfit_1.5-9.4
## [76] grid_3.6.2 blob_1.2.1 reprex_0.3.0
## [79] digest_0.6.27 xtable_1.8-4 openssl_1.4.3
## [82] munsell_0.5.0 askpass_1.1References
Nozue K, Devisetty UK, Lekkala S, Mueller-Moule P, Bak A, Casteel CL, Maloof JN (2018) Network analysis reveals a role for salicylic acid pathway components in shade avoidance. Plant Physiol. doi: 10.1104/pp.18.00920↩