purpose
Over-representation analysis (“enrichment analysis”) of RNA-seq data considering cDNA length effects with unsupported model organisms.
background
Among many transcriptome analysis platforms, transcriptome analysis by next-generation sequencing (RNA-seq) has been used widely among model and non-model organisms. In RNA-seq, number of reads mapped to reference cDNA sequences (or genes in genome sequences) is proportional to size of genes1. This effect should be corrected especially when enrichment analysis is performed because a gene category of your interest is enriched only because genes in the category are large in size. R/Bioconductor package GOseq handles this issue and I’ve been using it for my analysis2 . In my “Over-representation analysis” blog series, I would like to describe how to use the “GOseq” in model organmisms (such as Arabidopsis thaliana as a model plant) using GO terms (part1), or custom categories defied by users (part2), and non-model organisms in which a fasta file for cDNAs is available (part3). Furthermore, visualization of the enrichment summary in a heatmap will be described in part4 (instead of long tables!). For details in goseq, please read the origianl GOseq paper and papers cited the papers in PubMed.
# knitr::opts_chunk$set(echo = FALSE, error=TRUE)
library(goseq)
library(tidyverse)
library(readxl)GOseq analysis of Arabidopsis thaliana DEGs
Please download shade responsive genes Nozue (2018) PloS Genetics S2 Table1. Also download normalized read count file from the repository (click “Raw” button)

download myRNAseq.cpm from repository
Obtaining Arabidopsis thaliana DEGs (results=‘hide’)
# Unfortunately Arabidopsis thaliana was not supported by goseq.
head(supportedOrganisms())## Genome Id Id Description Lengths in geneLeneDataBase
## 12 anoCar1 ensGene Ensembl gene ID TRUE
## 13 anoGam1 ensGene Ensembl gene ID TRUE
## 14 apiMel2 ensGene Ensembl gene ID TRUE
## 56 bosTau2 geneSymbol Gene Symbol TRUE
## 15 bosTau3 ensGene Ensembl gene ID TRUE
## 57 bosTau3 geneSymbol Gene Symbol TRUE
## GO Annotation Available
## 12 FALSE
## 13 TRUE
## 14 FALSE
## 56 TRUE
## 15 TRUE
## 57 TRUE# reading shade-responsive genes (1h shade treatent)
getwd()## [1] "/Volumes/data_personal/Kazu_blog14/content/post/2018-09-26-over-representation-analysis-1-goseq-with-go-terms-in-unsupported-model-organisms"list.files(file.path("..","..",".."))## [1] "config.yaml" "content"
## [3] "data" "index.Rmd"
## [5] "Kazu_blog" "Kazu_blog14.Rproj"
## [7] "layouts" "netlify.toml"
## [9] "R" "resources"
## [11] "Slide-with-Xarigan.html" "static"
## [13] "themes"genes.shade1h<-readxl::read_excel(file.path("..","..","..","resources","2018-09-26-over-representation-analysis-1-goseq-with-arabidopsis-go-term_files","journal.pgen.1004953.s008.XLSX"),sheet=1)
genes.shade1h.up <- genes.shade1h %>% filter(table.logFC>0 & table.FDR<1.0e-15) # Up regulated by shade. Play with table.FDR
head(genes.shade1h.up)## # A tibble: 6 x 9
## AGI Description table.logConc table.logFC table.PValue table.FDR
## <chr> <chr> <dbl> <dbl> <dbl> <dbl>
## 1 AT4G… Encodes an… 33.0 34.1 0 0
## 2 AT3G… basic heli… 19.2 7.33 0 0
## 3 AT4G… Auxin indu… 16.0 4.86 0 0
## 4 AT5G… Encodes a … 16.6 4.15 0 0
## 5 AT3G… Primary au… 15.5 3.51 0 0
## 6 AT5G… SAUR-like … 17.3 3.47 0 0
## # … with 3 more variables: `found in ATH1` <chr>, Kozuka_2010_leaf_blade <dbl>,
## # Kozuka_2010_petiole <dbl>genes.shade1h.down <- genes.shade1h %>% filter(table.logFC>0 & table.FDR<1.0e-15) # Down regualated by shade. Play with table.FDR
head(genes.shade1h.down)## # A tibble: 6 x 9
## AGI Description table.logConc table.logFC table.PValue table.FDR
## <chr> <chr> <dbl> <dbl> <dbl> <dbl>
## 1 AT4G… Encodes an… 33.0 34.1 0 0
## 2 AT3G… basic heli… 19.2 7.33 0 0
## 3 AT4G… Auxin indu… 16.0 4.86 0 0
## 4 AT5G… Encodes a … 16.6 4.15 0 0
## 5 AT3G… Primary au… 15.5 3.51 0 0
## 6 AT5G… SAUR-like … 17.3 3.47 0 0
## # … with 3 more variables: `found in ATH1` <chr>, Kozuka_2010_leaf_blade <dbl>,
## # Kozuka_2010_petiole <dbl>check expression pattern of DEGs results=‘hide’
# load read count data from downloaded repository
# myRNAseq.cpm<-read_csv("resources/knozue-sasphenotyping-b04217e6559b/SAS_analysis_Jan2014/R_scripts_data_objects/myRNAseq.cpm.csv") # change path for your file
# Another way is to load read count data from downloaded csv file
#myRNAseq.cpm<-read_csv("2018-09-26-over-representation-analysis-1-goseq-with-arabidopsis-go-term_files/myRNAseq.cpm.csv")
# Read directly from repository
myRNAseq.cpm <- read_csv("https://bitbucket.org/knozue/sasphenotyping/raw/b04217e6559bf953fb03b67071d793c23f112495/SAS_analysis_Jan2014/R_scripts_data_objects/myRNAseq.cpm.csv")
# format data for plot
# genes.shade1h.up %>% left_join(myRNAseq.cpm,by=c("AGI"="X1")) %>% separate(Description,into="Description",sep=";",drop=TRUE) %>% unite(AGI_desc,c("AGI","Description")) %>% dplyr::select(-starts_with("table"),-`found in ATH1`,-starts_with("Kozuka")) -> plot.data
#
genes.shade1h.up %>% left_join(myRNAseq.cpm,by=c("AGI"="X1")) %>% separate(Description,into="Description",sep=";",extra="drop") %>% unite(AGI_desc,c("AGI","Description")) %>% dplyr::select(-starts_with("table"),-`found in ATH1`,-starts_with("Kozuka")) -> plot.data
plot.data## # A tibble: 20 x 9
## AGI_desc Col_Shade_1_rep… Col_Shade_1_rep… Col_Sun_1_repli… Col_Sun_1_repli…
## <chr> <dbl> <dbl> <dbl> <dbl>
## 1 "AT4G37… 17.7 14.1 0 0
## 2 "AT3G21… 29.0 12.4 0.249 0
## 3 "AT4G32… 84.8 75.3 2.49 3.07
## 4 "AT5G07… 41.3 42.8 2.49 2.23
## 5 "AT3G15… 88.9 56.9 7.97 4.74
## 6 "AT5G18… 18.2 23.0 0.747 3.07
## 7 "AT5G52… 90.0 60.9 6.48 10.3
## 8 "AT5G02… 58.2 30.2 5.23 6.14
## 9 "AT5G46… 85.2 86.2 11.0 11.4
## 10 "AT4G16… 42.0 43.1 2.99 8.37
## 11 "AT5G66… 70.7 86.8 12.0 11.2
## 12 "AT5G12… 113. 90.5 15.9 16.2
## 13 "AT1G21… 39.8 35.1 5.48 6.70
## 14 "AT5G02… 156. 151. 27.1 24.3
## 15 "AT3G23… 142. 120. 24.7 25.1
## 16 "AT1G26… 152. 164. 25.4 36.0
## 17 "AT5G47… 77.2 80.8 14.4 17.0
## 18 "AT4G27… 89.8 67.0 15.2 18.1
## 19 "AT4G25… 110. 110. 21.9 25.7
## 20 "AT1G67… 278. 242. 59.0 69.8
## # … with 4 more variables: Col_Shade_4_replicate1 <dbl>,
## # Col_Shade_4_replicate2 <dbl>, Col_Sun_4_replicate1 <dbl>,
## # Col_Sun_4_replicate2 <dbl>plot: use labeller = label_wrap_gen(width=10) for multiple line in each gene name
#p <- plot.data %>% gather(sample,value,-1) %>% separate(sample,c("genotype","trt","time_hr","rep"),sep="_") %>%
#ggplot(aes(x=fct_relevel(trt,"Sun"),y=value,color=trt,shape=time_hr)) + geom_boxplot() + geom_jitter() + facet_wrap(AGI_desc~.,scale="free",labeller = label_wrap_gen(width=30),ncol=3)+theme(strip.text = element_text(size=6))
# gather into pivod_longer
p <- plot.data %>% pivot_longer(-1,names_to="sample",values_to="value") %>% separate(sample,c("genotype","trt","time_hr","rep"),sep="_") %>%
ggplot(aes(x=fct_relevel(trt,"Sun"),y=value,color=trt,shape=time_hr)) + geom_boxplot() + geom_jitter() + facet_wrap(AGI_desc~.,scale="free",labeller = label_wrap_gen(width=30),ncol=3)+theme(strip.text = element_text(size=6))
p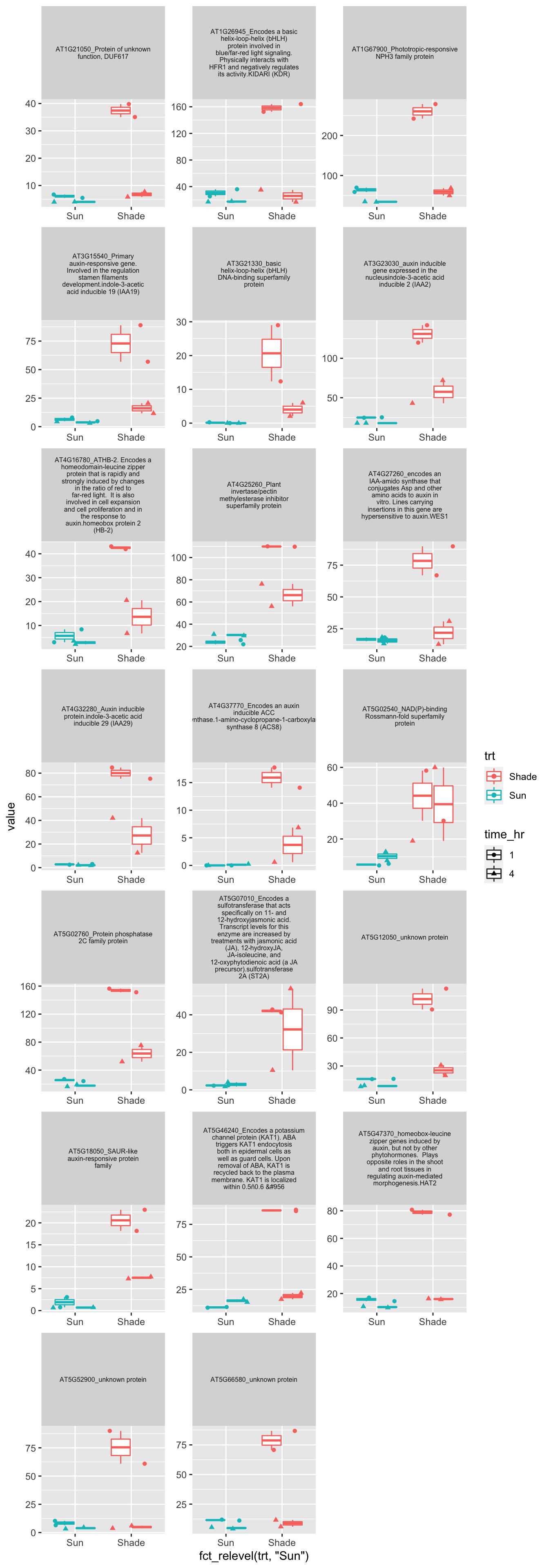
Usage of GOseq.ORA function
getwd()## [1] "/Volumes/data_personal/Kazu_blog14/content/post/2018-09-26-over-representation-analysis-1-goseq-with-go-terms-in-unsupported-model-organisms"# GOseq
library(ShortRead);library(goseq);library(GO.db);library("annotate")## Warning: package 'DelayedArray' was built under R version 3.6.3# Download cDNA sequence file from [Araport11] (https://www.arabidopsis.org/download_files/Sequences/Araport11_blastsets/Araport11_genes.201606.cdna.fasta.gz)
# At_cdna<-Biostrings::readDNAStringSet("2018-09-26-over-representation-analysis-1-goseq-with-arabidopsis-go-term_files/Araport11_genes.201606.cdna.fasta")
# donot use Araport11 version due to multiple splicing variants
# select only representative splicing variant
# Download cDNA sequence file (representative) from [TAIR10](https://www.arabidopsis.org/download_files/Sequences/TAIR10_blastsets/TAIR10_cdna_20110103_representative_gene_model_updated)
#system("gunzip -c 2018-09-26-over-representation-analysis-1-goseq-with-arabidopsis-go-term_files/TAIR10_cdna_20110103_representative_gene_model.gz > 2018-09-26-over-representation-analysis-1-goseq-with-arabidopsis-go-term_files/TAIR10_cdna_20110103_representative_gene_model")
#At_cdna<-Biostrings::readDNAStringSet("2018-09-26-over-representation-analysis-1-goseq-with-arabidopsis-go-term_files/TAIR10_cdna_20110103_representative_gene_model")
# another way of download files
#system("wget https://www.arabidopsis.org/download_files/Sequences/TAIR10_blastsets/TAIR10_cds_20110103_representative_gene_model_updated") # error
download.file(url="https://www.arabidopsis.org/download_files/Sequences/TAIR10_blastsets/TAIR10_cds_20110103_representative_gene_model_update",destfile="../../../resources/2018-09-26-over-representation-analysis-1-goseq-with-arabidopsis-go-term_files/TAIR10_cdna_20110103_representative_gene_model.gz") # error## Warning in download.file(url = "https://www.arabidopsis.org/
## download_files/Sequences/TAIR10_blastsets/
## TAIR10_cds_20110103_representative_gene_model_update", : URL 'https://
## www.arabidopsis.org/download_files/Sequences/TAIR10_blastsets/
## TAIR10_cds_20110103_representative_gene_model_update': status was 'Peer
## certificate cannot be authenticated with given CA certificates'## Error in download.file(url = "https://www.arabidopsis.org/download_files/Sequences/TAIR10_blastsets/TAIR10_cds_20110103_representative_gene_model_update", : cannot open URL 'https://www.arabidopsis.org/download_files/Sequences/TAIR10_blastsets/TAIR10_cds_20110103_representative_gene_model_update'download directly from TAIR
- Works only from campus network
At_cdna <- Biostrings::readDNAStringSet("https://www.arabidopsis.org/download_files/Sequences/TAIR10_blastsets/TAIR10_cds_20110103_representative_gene_model_updated") # In addition: Warning message:In download.file(fp, filepath2[i]) : URL 'https://www.arabidopsis.org/download_files/Sequences/TAIR10_blastsets/TAIR10_cds_20110103_representative_gene_model_updated': status was 'Peer certificate cannot be authenticated with given CA certificates' (032121). NO error from on campusUsing downloaded file (TAIR10_cdna_20110103_representative_gene_model_updated.gz)
getwd()## [1] "/Volumes/data_personal/Kazu_blog14/content/post/2018-09-26-over-representation-analysis-1-goseq-with-go-terms-in-unsupported-model-organisms"system("gunzip -c ../../../resources/2018-09-26-over-representation-analysis-1-goseq-with-arabidopsis-go-term_files/TAIR10_cdna_20110103_representative_gene_model_updated.gz > ../../../resources/2018-09-26-over-representation-analysis-1-goseq-with-arabidopsis-go-term_files/TAIR10_cdna_20110103_representative_gene_model_updated")
At_cdna <- Biostrings::readDNAStringSet(file.path("..","..","..","resources","2018-09-26-over-representation-analysis-1-goseq-with-arabidopsis-go-term_files","TAIR10_cdna_20110103_representative_gene_model_updated"))
At_cdna## A DNAStringSet instance of length 33602
## width seq names
## [1] 2394 AGAAAACAGTCGACCGTCATT...TATTTGGTAATTTTTTGAGTC AT1G50920.1 | Sym...
## [2] 546 ATGACTCGTTTGTTGCCTTAC...CAAGTTGATTCTGGTACATAG AT1G36960.1 | Sym...
## [3] 2314 ATGGATTCAGAGTCAGAGTCA...CCTGGTGCATTGTGTTTCTCC AT1G44020.1 | Sym...
## [4] 1658 TCGTTTCGTCGTCGATCAGAA...ATCGATTACATGCTACATTTT AT1G15970.1 | Sym...
## [5] 1453 ATTGAAAAGAAAACACATCCC...GACCACCAAAATCTTCTCATA AT1G73440.1 | Sym...
## ... ... ...
## [33598] 87 GGATGGATGTCTGAGCGGTTG...GTTCGAATCCCTCTCCATCCG ATMG00420.1 | Sym...
## [33599] 384 ATGCTCCCCGCCGGTTGTTGG...CTACGATACTTAACTATATAA ATMG01330.1 | Sym...
## [33600] 573 ATGGATAACCAATTCATTTTC...GAACAGCGTAGCGACGGATAA ATMG00070.1 | Sym...
## [33601] 366 ATGGCATCAAAAATCCGCAAA...GATCCTTCTGCATACGCATAA ATMG00130.1 | Sym...
## [33602] 74 GCGCTCTTAGTTCAGTTCGGT...GTTCAAATCCTACAGAGCGTG ATMG00930.1 | Sym...# remove uncompressed file
system("rm ../../../resources/2018-09-26-over-representation-analysis-1-goseq-with-arabidopsis-go-term_files/TAIR10_cdna_20110103_representative_gene_model_updated")Run this chunck once to make Atgoslim.TAIR.BP.list.Rdata
## download TAIR version of [GOSLIM](http://www.arabidopsis.org/download/index-auto.jsp?dir=%2Fdownload_files%2FGO_and_PO_Annotations%2FGene_Ontology_Annotations)
## read me file is http://www.arabidopsis.org/download_files/GO_and_PO_Annotations/Gene_Ontology_Annotations/ATH_GO.README.txt
## directly read the file from TAIR
#test<-readr::read_tsv("https://www.arabidopsis.org/download_files/Subscriber_Data_Releases/TAIR_Data_20180630/ATH_GO_GOSLIM.txt.gz",skip=2,col_names=FALSE) # does not work
## store uncompressed version in box.com and download it
#test2<-readr::read_tsv("https://app.box.com/s/pdxbg2t8vpyatp4x9eewc13qaloa5i4r",skip=2,col_names=FALSE) # does not work.
## download file
#download.file("https://www.arabidopsis.org/download_files/Subscriber_Data_Releases/TAIR_Data_20180630/ATH_GO_GOSLIM.txt.gz",destfile ="/../../../resources/2018-09-26-over-representation-analysis-1-goseq-with-arabidopsis-go-term_files/ATH_GO_GOSLIM.txt.gz")
# updated (032221, the link was not found)
#download.file("https://www.arabidopsis.org/download_files/GO_and_PO_Annotations/Gene_Ontology_Annotations/ATH_GO_GOSLIM.txt.gz",destfile ="../../../resources/2018-09-26-over-representation-analysis-1-goseq-with-arabidopsis-go-term_files/ATH_GO_GOSLIM.txt.gz") # does not work
system("unzip -c ../../../resources/2018-09-26-over-representation-analysis-1-goseq-with-arabidopsis-go-term_files/ATH_GO_GOSLIM.txt_downloaded032221.gz > ../../../resources/2018-09-26-over-representation-analysis-1-goseq-with-arabidopsis-go-term_files/ATH_GO_GOSLIM.txt") # this is not gunzip.
Atgoslim.TAIR <- read_tsv(file.path("..","..","..","resources","2018-09-26-over-representation-analysis-1-goseq-with-arabidopsis-go-term_files","ATH_GO_GOSLIM.txt"),skip=6,col_names = FALSE) # previous version needs skip=2##
## ── Column specification ────────────────────────────────────────────────────────
## cols(
## X1 = col_character(),
## X2 = col_character(),
## X3 = col_character(),
## X4 = col_character(),
## X5 = col_character(),
## X6 = col_character(),
## X7 = col_double(),
## X8 = col_character(),
## X9 = col_character(),
## X10 = col_character(),
## X11 = col_character(),
## X12 = col_character(),
## X13 = col_character(),
## X14 = col_character(),
## X15 = col_date(format = "")
## )Atgoslim.TAIR %>% filter(X8=="P",X1=="AT4G38360") %>% dplyr::select(1,5,6,8,9,10)## # A tibble: 15 x 6
## X1 X5 X6 X8 X9 X10
## <chr> <chr> <chr> <chr> <chr> <chr>
## 1 AT4G38… transport GO:000… P transport IBA
## 2 AT4G38… vesicle-mediated transport to t… GO:009… P other cellular … IGI
## 3 AT4G38… vesicle-mediated transport to t… GO:009… P transport IGI
## 4 AT4G38… negative regulation of brassino… GO:190… P response to che… IGI
## 5 AT4G38… negative regulation of brassino… GO:190… P response to end… IGI
## 6 AT4G38… plant-type hypersensitive respo… GO:000… P response to str… IEA
## 7 AT4G38… plant-type hypersensitive respo… GO:000… P response to bio… IEA
## 8 AT4G38… negative regulation of brassino… GO:190… P other cellular … IGI
## 9 AT4G38… plant-type hypersensitive respo… GO:000… P other cellular … IEA
## 10 AT4G38… negative regulation of brassino… GO:190… P signal transduc… IGI
## 11 AT4G38… programmed cell death GO:001… P cell death IMP
## 12 AT4G38… negative regulation of brassino… GO:190… P cell communicat… IGI
## 13 AT4G38… vacuole organization GO:000… P cellular compon… IGI
## 14 AT4G38… plant-type hypersensitive respo… GO:000… P cell death IEA
## 15 AT4G38… plant-type hypersensitive respo… GO:000… P response to ext… IEAAtgoslim.TAIR.BP <-Atgoslim.TAIR%>% filter(X8=="P")
Atgoslim.TAIR.BP.list<-tapply(Atgoslim.TAIR.BP$X6,Atgoslim.TAIR.BP$X1,c)
save(Atgoslim.TAIR.BP.list,file=file.path("..","..","..","resources","2018-09-26-over-representation-analysis-1-goseq-with-arabidopsis-go-term_files","Atgoslim.TAIR.BP.list.Rdata"))
system("rm /../../../resources/2018-09-26-over-representation-analysis-1-goseq-with-arabidopsis-go-term_files/ATH_GO_GOSLIM.txt*") # remove both filesGOseq.Atgoslim.BP.list.ORA function
This function is used to calculate GO ORA results multiple times.
load("../../../resources/2018-09-26-over-representation-analysis-1-goseq-with-arabidopsis-go-term_files/Atgoslim.TAIR.BP.list.Rdata")
head(Atgoslim.TAIR.BP.list)## $`18S RRNA`
## [1] "GO:0006412"
##
## $`25S RRNA`
## [1] "GO:0043043" "GO:0043043" "GO:0043043"
##
## $`5.8S RRNA`
## [1] "GO:0043043" "GO:0043043" "GO:0043043"
##
## $AAN
## [1] "GO:0007623"
##
## $AAR1
## [1] "GO:0006970" "GO:0006970" "GO:0040029"
##
## $AAR2
## [1] "GO:0009845" "GO:0040029"GOseq.Atgoslim.BP.list.ORA<-function(genelist,padjust=0.05,ontology="BP",custom.category.list=Atgoslim.TAIR.BP.list,cdna=At_cdna) { # return GO enrichment table, padjus, padjust=0.05.
bias<-nchar(cdna)
names(bias) <- tibble(AGI=names(cdna)) %>% separate(AGI,into=c("AGI2" ,"test1","test2"),sep=" | ",extra="drop") %>% separate(AGI2,into="AGI3",sep="\\.",extra="drop") %>% dplyr::select(AGI3) %>% as_vector()
table(duplicated(names(bias))) # revised (032221)
#TF<-(names(bias) %in% genelist)*1
TF<-as.integer(names(bias) %in% genelist)
names(TF)<-names(bias)
#print(TF)
pwf<-nullp(TF,bias.data=bias)
#print(pwf$DEgenes)
GO.pval <- goseq(pwf,gene2cat=custom.category.list,use_genes_without_cat=TRUE) # format became different in new goseq version (021111). Does not work (042716)
#head(GO.pval)
if(ontology=="BP") {
GO.pval2<-subset(GO.pval,ontology=="BP")
} else if(ontology=="CC") {
GO.pval2<-subset(GO.pval,ontology=="CC")
} else {
GO.pval2<-subset(GO.pval,ontology=="MF")
}
# calculating padjust by BH
GO.pval2$over_represented_padjust<-p.adjust(GO.pval2$over_represented_pvalue,method="BH")
if(GO.pval2$over_represented_padjust[1]>padjust) return("no enriched GO")
else {
enriched.GO<-GO.pval2[GO.pval2$over_represented_padjust<padjust,]
print("enriched.GO is")
print(enriched.GO)
## write Term and Definition
for(i in 1:dim(enriched.GO)[1]) {
if(is.null(Term(GOTERM[enriched.GO[i,"category"]]))) {next} else {
enriched.GO$Term[i]<-Term(GOTERM[[enriched.GO[i,"category"]]])
enriched.GO$Definition[i]<-Definition(GOTERM[[enriched.GO[i,"category"]]])
}
}
return(enriched.GO)
}
}GO.seq
# genes.shade1h.up
genes.shade1h.up$AGI## [1] "AT4G37770" "AT3G21330" "AT4G32280" "AT5G07010" "AT3G15540" "AT5G18050"
## [7] "AT5G52900" "AT5G02540" "AT5G46240" "AT4G16780" "AT5G66580" "AT5G12050"
## [13] "AT1G21050" "AT5G02760" "AT3G23030" "AT1G26945" "AT5G47370" "AT4G27260"
## [19] "AT4G25260" "AT1G67900"GOseq.Atgoslim.BP.list.ORA(genelist=genes.shade1h.up$AGI)## Using manually entered categories.## Calculating the p-values...## 'select()' returned 1:1 mapping between keys and columns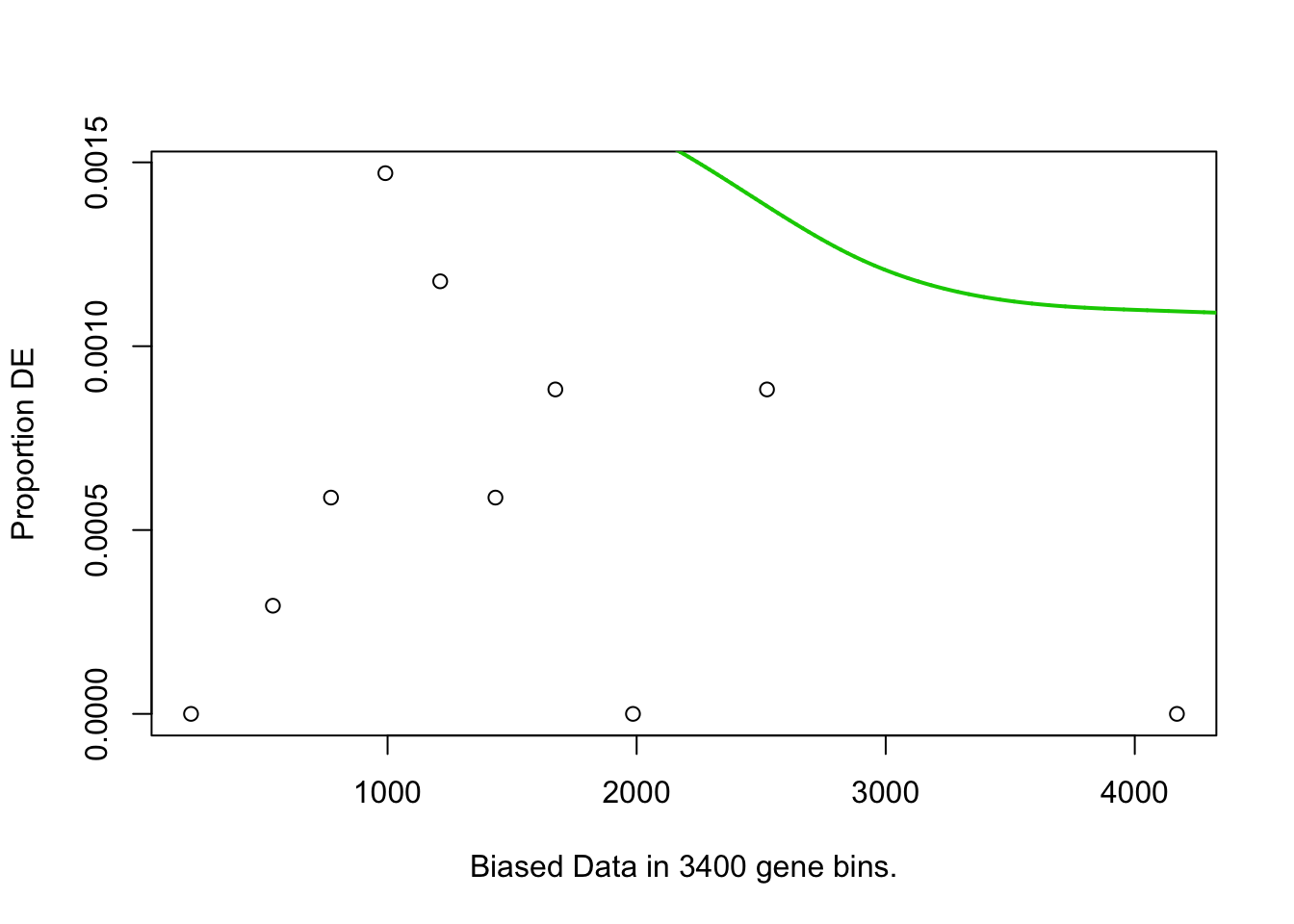
## [1] "enriched.GO is"
## category over_represented_pvalue under_represented_pvalue numDEInCat
## 981 GO:0009733 5.840352e-09 1 6
## 935 GO:0009641 1.325751e-07 1 3
## numInCat term ontology over_represented_padjust
## 981 251 response to auxin BP 2.312195e-05
## 935 17 shade avoidance BP 2.624325e-04## category over_represented_pvalue under_represented_pvalue numDEInCat
## 981 GO:0009733 5.840352e-09 1 6
## 935 GO:0009641 1.325751e-07 1 3
## numInCat term ontology over_represented_padjust
## 981 251 response to auxin BP 2.312195e-05
## 935 17 shade avoidance BP 2.624325e-04
## Term
## 981 response to auxin
## 935 shade avoidance
## Definition
## 981 Any process that results in a change in state or activity of a cell or an organism (in terms of movement, secretion, enzyme production, gene expression, etc.) as a result of an auxin stimulus.
## 935 Shade avoidance is a set of responses that plants display when they are subjected to the shade of another plant. It often includes elongation, altered flowering time, increased apical dominance and altered partitioning of resources. Plants are able to distinguish between the shade of an inanimate object (e.g. a rock) and the shade of another plant due to the altered balance between red and far-red light in the shade of a plant; this balance between red and far-red light is perceived by phytochrome.see R scripts for vignett
Session info
sessionInfo()## R version 3.6.2 (2019-12-12)
## Platform: x86_64-apple-darwin15.6.0 (64-bit)
## Running under: macOS Mojave 10.14.6
##
## Matrix products: default
## BLAS: /Library/Frameworks/R.framework/Versions/3.6/Resources/lib/libRblas.0.dylib
## LAPACK: /Library/Frameworks/R.framework/Versions/3.6/Resources/lib/libRlapack.dylib
##
## locale:
## [1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
##
## attached base packages:
## [1] parallel stats4 stats graphics grDevices utils datasets
## [8] methods base
##
## other attached packages:
## [1] annotate_1.64.0 XML_3.99-0.3
## [3] GO.db_3.10.0 AnnotationDbi_1.48.0
## [5] ShortRead_1.44.3 GenomicAlignments_1.22.1
## [7] SummarizedExperiment_1.16.1 DelayedArray_0.12.3
## [9] matrixStats_0.57.0 Biobase_2.46.0
## [11] Rsamtools_2.2.3 Biostrings_2.54.0
## [13] XVector_0.26.0 BiocParallel_1.20.1
## [15] rtracklayer_1.46.0 GenomicRanges_1.38.0
## [17] GenomeInfoDb_1.22.1 IRanges_2.20.2
## [19] S4Vectors_0.24.4 BiocGenerics_0.32.0
## [21] readxl_1.3.1 forcats_0.5.0
## [23] stringr_1.4.0 dplyr_1.0.2
## [25] purrr_0.3.4 readr_1.4.0
## [27] tidyr_1.1.2 tibble_3.0.4
## [29] ggplot2_3.3.3 tidyverse_1.3.0
## [31] goseq_1.38.0 geneLenDataBase_1.22.0
## [33] BiasedUrn_1.07
##
## loaded via a namespace (and not attached):
## [1] colorspace_2.0-0 hwriter_1.3.2 ellipsis_0.3.1
## [4] fs_1.5.0 rstudioapi_0.13 farver_2.0.3
## [7] bit64_4.0.5 fansi_0.4.1 lubridate_1.7.9.2
## [10] xml2_1.3.2 splines_3.6.2 knitr_1.31
## [13] jsonlite_1.7.2 broom_0.7.3 dbplyr_2.0.0
## [16] png_0.1-7 compiler_3.6.2 httr_1.4.2
## [19] backports_1.2.1 assertthat_0.2.1 Matrix_1.3-2
## [22] cli_2.2.0 htmltools_0.5.1.1 prettyunits_1.1.1
## [25] tools_3.6.2 gtable_0.3.0 glue_1.4.2
## [28] GenomeInfoDbData_1.2.2 rappdirs_0.3.1 Rcpp_1.0.6
## [31] cellranger_1.1.0 vctrs_0.3.6 nlme_3.1-151
## [34] blogdown_1.2.2 xfun_0.22 rvest_0.3.6
## [37] lifecycle_0.2.0 zlibbioc_1.32.0 scales_1.1.1
## [40] hms_0.5.3 RColorBrewer_1.1-2 yaml_2.2.1
## [43] curl_4.3 memoise_1.1.0 biomaRt_2.42.1
## [46] latticeExtra_0.6-29 stringi_1.5.3 RSQLite_2.2.1
## [49] highr_0.8 GenomicFeatures_1.38.2 rlang_0.4.10
## [52] pkgconfig_2.0.3 bitops_1.0-6 evaluate_0.14
## [55] lattice_0.20-41 labeling_0.4.2 bit_4.0.4
## [58] tidyselect_1.1.0 magrittr_2.0.1 bookdown_0.21
## [61] R6_2.5.0 generics_0.1.0 DBI_1.1.0
## [64] pillar_1.4.7 haven_2.3.1 withr_2.3.0
## [67] mgcv_1.8-33 RCurl_1.98-1.2 modelr_0.1.8
## [70] crayon_1.3.4 utf8_1.1.4 BiocFileCache_1.10.2
## [73] rmarkdown_2.7 jpeg_0.1-8.1 progress_1.2.2
## [76] grid_3.6.2 blob_1.2.1 reprex_0.3.0
## [79] digest_0.6.27 xtable_1.8-4 openssl_1.4.3
## [82] munsell_0.5.0 askpass_1.1References